- Suppliers






ADM 
Antares 
Armor Pharma 
Asahi KASEI 
Ashland 
BASF 
Beneo 
Captisol 
Clariant 
Croda 
DFE Pharma 
Dow 
Excipio Chemicals 
Fuji Chemical 
Gattefossé 
Gangwal 
IOI Oleo 
Ingredient Pharm 
JRS Pharma 
Kerry 
KLK Oleo 
Lipoid 
Lubrizol Life Science Health 
MAGNESIA 
MEGGLE 
Nagase Viita 
Nordic Bioproducts 
pharm-a-spheres 
PMC Isochem 
PQ 
Seppic 
ShinEtsu 
Sigachi 
SPI Pharma 
Südzucker 
Vikram Thermo 
Zerion Pharma  All4Nutra
All4Nutra
- Suppliers
ADM Antares Armor Pharma Asahi KASEI Ashland BASF Beneo Captisol Clariant Croda DFE Pharma Dow Excipio Chemicals Fuji Chemical Gattefossé Gangwal IOI Oleo Ingredient Pharm JRS Pharma Kerry KLK Oleo Lipoid Lubrizol Life Science Health MAGNESIA MEGGLE Nagase Viita Nordic Bioproducts pharm-a-spheres PMC Isochem PQ Seppic ShinEtsu Sigachi SPI Pharma Südzucker Vikram Thermo Zerion Pharma - All4Nutra

Chinese Excipient DMF

DMF-Regulations-in-China - Graphic by FDLI
Since January 1, 2018, CFDA has not been accepting Active Pharmaceutical Ingredients (APIs), Pharmaceutical Excipients and Pharmaceutical Packaging Materials registration application independently. The Active Pharmaceutical Ingredients (APIs), Pharmaceutical Excipients and Pharmaceutical Packaging Materials are now reviewed/approved as part of a drug product application.
Up to now there are 2.324 activated Chinese DMF for excipients in the list of the CDE. In total there are 4.278 DMF numbers for excipients in the CDA database. Quite a significant amount in comparison to the number of US DMFs for excipients – see our updated list here.
The status of the Chinese DMF from “I” (Raw materials/excipients/packages that have not yet passed the co-review and approval with the formulation) to “A” (Raw materials/excipients/packages that are approved for use in marketed formulations) will automatically be changed once the drug registration which contains the excipient / API or packaging material is approved.
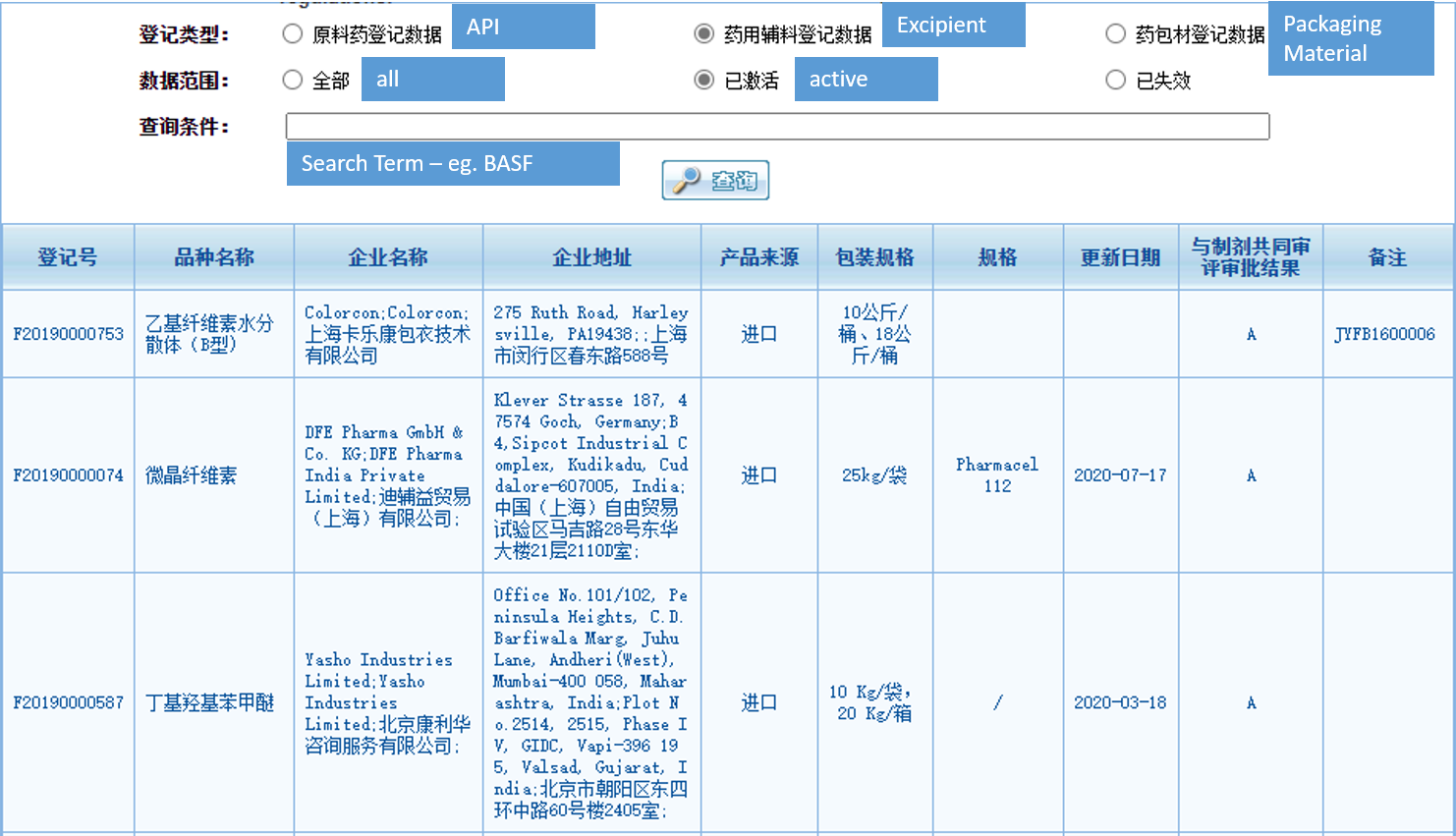
The list is online here: https://www.cde.org.cn/main/xxgk/listpage/ba7aed094c29ae31467c0a35463a716e
For an easier search we have translated the most important parts to search the list
More Foreign Drug Companies Registering Drug Master Files in China
By Ames Gross, President, Pacific Bridge Medical – FDLI Food and Drug Law Institute
Introduction
China’s drug market has exploded over the last few years, and future growth is estimated at 6-7% over the next five years. Valued at about $140 billion, it is now the second-largest drug market in the world. With such growth, there have also been positive regulatory reforms. Since China joined the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) in June 2017, key regulatory reforms include: 1) reduction of the Investigational New Drug (IND) clinical trial approval timelines from one to two years to sixty days; 2) creation of conditional approval for drugs that fulfill unmet needs; and 3) increased acceptance of foreign clinical data, among others. Opportunities for foreign drug and biotech firms in China have never been greater. In addition, Active Pharmaceutical Ingredient (API), excipient, and drug packaging manufacturers can also export their products and benefit from China’s strong drug market growth by filing Drug Master Files (DMFs) in China.
Regulatory Background of DMFs in China
The National Medical Products Administration (NMPA) was founded on the basis of the former China Food and Drug Administration (CFDA). In August 2016, to simplify the drug review procedure, the CFDA announced that the review and approval of APIs, excipients, and pharmaceutical packaging materials would be reviewed individually and would be approved with a registration license for pre-market approval. This expensive and time-consuming registration process was separate from drug approval. In November 2017, the CFDA changed their position and announced that the review and approval of APIs, excipients, and packaging materials would no longer be reviewed individually and approved with a separate registration license. CFDA published this change in the “Announcement on Adjusting the Review and Approval of a Drug Substance, Pharmaceutical Excipient, and Pharmaceutical Packaging Materials” on November 23, 2017.
On January 1, 2018, the NMPA announced that DMFs are required for the importation of APIs, excipients, and packaging materials. In July 2019, the NMPA announced that if the manufacturer cannot file a DMF for certain reasons, the technical dossiers must be submitted by the final drug manufacturer. A DMF is considered “inactive” after a DMF number is issued. A DMF is considered “active” only after an Administrative Review. Only an activated DMF can be imported for marketing purposes into China. A product with an inactive DMF can only be imported for research and development purposes.
Overseas API, excipient, or packaging material manufacturers must then authorize their domestic subsidiary or a China agent to file the DMF. Both the manufacturer and China agent are responsible for the authenticity and integrity of the filed DMF dossiers. The DMF holder will then authorize the final drug applicant to use their DMF for the drug registration. The drug applicant, or the market authorization holder, bears the major responsibility for the quality of the drug.
Steps of the DMF Filing Procedure
The first step is to appoint a local China agent or utilize the manufacturer’s subsidiary in China as the local agent. Because of the language differences and the ever-changing regulations in China, a reliable local agent is critical for an overseas manufacturer to deal with the complex regulatory issues in China. Clear correspondence is critical to the overall process and will help expedite the process.
The second step is putting together the required dossier. The local agent will provide a critical checklist to guide the collection of technical dossiers. The local agent or consultant from China will also prove appropriate templates and interpretations.
Third, the local agent will put together the registration dossiers. It may take three months for the compilation of the registration dossier and its translation from English to Chinese to be completed. Generally, the DMF number for excipient or packaging materials is released in about one to two weeks upon receipt by China’s Center for Drug Evaluation (CDE); for an API, it may take about two to four weeks. Of course, this could go longer if the registration dossier is not complete and the CDE requires more information.
Finally, the details of the DMF file are not reviewed closely until the file goes through Administrative Review. An annual report on the API, excipient, or packaging material must be submitted by the local China agent in the first quarter every year; otherwise, the DMF will be canceled. Continue reading here on Chinese DMF







