- Suppliers






ADM 
Antares 
Armor Pharma 
Asahi KASEI 
Ashland 
BASF 
Beneo 
Captisol 
Clariant 
Croda 
DFE Pharma 
Dow 
Excipio Chemicals 
Fuji Chemical 
Gattefossé 
Gangwal 
IOI Oleo 
Ingredient Pharm 
JRS Pharma 
Kerry 
KLK Oleo 
Lipoid 
Lubrizol Life Science Health 
MAGNESIA 
MEGGLE 
Nagase Viita 
Nordic Bioproducts 
pharm-a-spheres 
PMC Isochem 
PQ 
Seppic 
ShinEtsu 
Sigachi 
SPI Pharma 
Südzucker 
Vikram Thermo 
Zerion Pharma  All4Nutra
All4Nutra
- Suppliers
ADM Antares Armor Pharma Asahi KASEI Ashland BASF Beneo Captisol Clariant Croda DFE Pharma Dow Excipio Chemicals Fuji Chemical Gattefossé Gangwal IOI Oleo Ingredient Pharm JRS Pharma Kerry KLK Oleo Lipoid Lubrizol Life Science Health MAGNESIA MEGGLE Nagase Viita Nordic Bioproducts pharm-a-spheres PMC Isochem PQ Seppic ShinEtsu Sigachi SPI Pharma Südzucker Vikram Thermo Zerion Pharma - All4Nutra

Cyclodextrin: A prospective nanocarrier for the delivery of antibacterial agents against bacteria that are resistant to antibiotics
Cyclodextrin: A prospective nanocarrier for the delivery of antibacterial agents against bacteria that are resistant to antibiotics
Abstract
Supramolecular chemistry introduces us to the macrocyclic host cyclodextrin, which has a hydrophobic cavity. The hydrophobic cavity has a higher affinity for hydrophobic guest molecules and forms host-guest complexation with non-covalent interaction. Three significant cyclodextrin kinds are α-cyclodextrin, β-cyclodextrin, and γ-cyclodextrin. The most often utilized is β-cyclodextrin (β-CD). An effective weapon against bacteria that are resistant to antibiotics is cyclodextrin. Several different kinds of cyclodextrin nanocarriers (β-CD, HP-β-CD, Meth-β-CD, cationic CD, sugar-grafted CD) are utilized to enhance the solubility, stability, dissolution, absorption, bioavailability, and permeability of the antibiotics. Cyclodextrin also improves the effectiveness of antibiotics, antimicrobial peptides, metallic nanoparticles, and photodynamic therapy (PDT). Again, cyclodextrin nanocarriers offer slow-release properties for sustained-release formulations where steady-state plasma antibiotic concentration is needed for an extended time. A novel strategy to combat bacterial resistance is a stimulus (pH, ROS)-responsive antibiotics released from cyclodextrin carrier. Once again, cyclodextrin traps autoinducer (AI), a crucial part of bacterial quorum sensing, and reduces virulence factors, including biofilm formation. Cyclodextrin helps to minimize MIC in particular bacterial strains, keep antibiotic concentrations above MIC in the infection site and minimize the possibility of antibiotic and biofilm resistance. Sessile bacteria trapped in biofilms are more resistant to antibiotic therapy than bacteria in a planktonic form. Cyclodextrin also involves delivering antibiotics to biofilm and resistant bacteria to combat bacterial resistance.
Introduction
Numerous infectious disorders have been treated, and hundreds of antibacterial drugs have been made therapeutically available since Alexander Fleming’s discovery of penicillin in 1928. However, due to their widespread use, antimicrobial resistance—one of current medicine’s most critical issues—arises, reducing conventional therapy’s therapeutic effect [[1]]. The “Golden Age” of antibiotic discovery is said to have occurred between 1940 and 1962, when more than 100 antibiotics, mostly derived from naturally occurring bacteria, were discovered [[2]]. But the rise of microbial resistance to already-existing medicines has prompted the ongoing discovery of new antibiotics. First-generation penicillin could be used to treat Staphylococcus aureus (S. aureus), but within a year, penicillin-resistant S. aureus started to emerge. New methicillin-based drugs were subsequently created to treat S. aureus resistant to penicillin, although Methicillin-resistant Staphylococcus aureus (MRSA) first appeared in 1986 [[3]]. The newly created vancomycin was crucial in the management of MRSA. However, vancomycin usage led to the development of vancomycin-resistant S. aureus (VRSA) [[3]]. Because of this, using recently created antibiotics may hasten the appearance of new antibiotic-resistant bacteria. It also implies that the likelihood of developing antibiotic-resistant bacteria increases with increasing antibiotic overuse [[4]]. The biggest threat to a patient’s condition is hospital-acquired bacterial infections, often brought on by nosocomial pathogens such as Pseudomonas aeruginosa, Acinetobacter baumannii, Staphylococcus aureus, Escherichia coli, Klebsiella pneumoniae, and others [[5]].
Recently developed community-acquired MRSA (CA-MRSA) variants quickly establish themselves as the community’s predominant pathogens [[6]]. Switching to more expensive, broad-spectrum antibiotics has become necessary due to the persistence of resistant strains and the continuously high rates of antibiotic usage in hospitals, communities, and agriculture [[7]]. Antibiotic resistance among bacteria that risk human health has been caused by this misuse and abuse of antibiotics [[8]], which has resulted in treatment problems and elevated healthcare costs [[7]]. Once they start feeling better, many patients tend to stop their treatments, which can worsen antibiotic resistance. Additionally, many antibiotics have short half-lives and require frequent administration, promoting compliance to the patient. The lack of compliance frequently causes treatments to fail or raises the expense of healthcare resources by requiring additional drugs and hospital admission [[9]]. Increased livestock production results from rising demand for animal protein, which increases the use of antibiotics in the agricultural and livestock sectors and, eventually, breeds antibiotic resistance [[10]]. Antibiotic resistance is complex. It is known that horizontal transmission of genes on plasmids or transposons, as well as spontaneous gene mutation, are the two ways bacteria develop resistance to both single and multiple antibiotics [[11]]. The resistant genes to the drug include blaZ, mecA, parC, gyrA, gyrB, sulA, drfB, ermA, ermb, ermc, vat, and vatB [[12]]. The expression of the resistance gene results in the capacity to fight antibiotics by a variety of processes, including (a) altering the drug’s drug targets, (b) degrading the antibiotic enzymatically, (c) reducing cell membrane permeability, and (d) efflux pumps [[13]]. Planktonic bacterial infections present acute threats and are getting harder and harder to cure as acquired antibiotic resistance rates rise. This problem is made more difficult when bacteria develop biofilms linked to recurrent and chronic bacterial infections [[14]]. Bacterial biofilms spur chronic infections because they are more resistant to phagocytosis and other defensive mechanisms, including phagocytosis and disinfectant chemicals [[15]]. Pathogenic biofilms are produced by populations of pathogens that attach to surfaces and are immersed in the extracellular matrix, including Pseudomonas aeruginosa, Staphylococcus aureus, Vibrio cholerae, and Nontuberculous mycobacteria [16, 17, 18].
The development of bacterial biofilms is one of the core aspects causing bacterial resistance [[19]]. The resistance of bacteria in biofilms to antibiotics can be up to 1000 times higher than planktonic bacteria [[20]]. Subinhibitory concentrations, which are antibiotic exposure levels below the minimum inhibitory concentration (MIC), might cause bacteria to be more capable of forming biofilms, which can reduce their sensitivity to antibiotics [[21]]. For example, it has recently been demonstrated that clinical Enterococcus faecalis isolates can develop biofilms more quickly when exposed to subinhibitory antibiotic doses [[22]]. The conventional antibiotics used for antibacterial therapy have certain drawbacks in contemporary medicine, such as limited bioavailability, little penetration into the infection nidus, and the emergence of drug-resistant bacteria (superbugs) [[23]]. Antibiotic resistance is currently being combated using various methods, including cationic polymers, nanoparticles, combinatorial therapy, antimicrobial photodynamic therapy (PDT), interference in bacterial quorum sensing, antimicrobial peptides, and chemical modification of antibiotics. By serving as either intrinsic therapies or nanocarriers for antimicrobial drugs, recent advancements in nanomaterial-based systems offer new ways to fight multi-drug-resistant planktonic and biofilm infections [[24]]. When used as antibacterial agents or as carriers for antibacterial drug loading, nanoparticles can increase the bioavailability and efficacy of antibiotics [[25]]. Compared to conventional therapy, the effectiveness of medication delivery using nano-systems increases while potential toxicity decreases. Because of their great attraction to bacteria, high specific surface area ratio, possibility for surface functionalization, and capacity to load drug molecules, nanoparticles play an important role in efficient antibacterial action [[26]]. Nanoparticles can shield medicines from enzyme attacks, prolong medication release, and boost half-life and bioavailability [[27]].
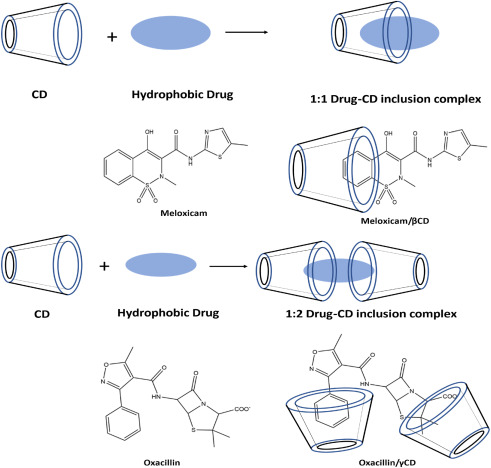
A successful strategy to raise the therapeutic index is the active targeting of nanoparticles at bacteria [[28]]. Reversible non-covalent interactions, a key component of supramolecular chemistry, such as electrostatic interactions, p-p stacking interactions, hydrophobic effects, van der Waals interactions, and hydrogen-bonding interactions, can successfully bring together like molecules through molecular recognition to create ordered supramolecular nanoarchitecture’s with adjustable sizes, morphology, and functions, such as micelles, nanoparticles (NPs), vesicles, and hydrogels [29, 30, 31]. Cyclodextrin (CD) is a supramolecular macrocyclic host, also known as α-, β- or γ-cyclodextrins, are cyclic oligosaccharides made up of various numbers of glucose molecules [[32]] and include six, seven, or eight glucose monomers connected by α-1,4-glucose linkages [[33]]. Due to their capacity to modify the physical, chemical, and biological characteristics of guest molecules through the creation of inclusion (host-guest) complexes (Fig. 1), cyclodextrins are prospective candidates for drug carriers [[34]]. Based on hydrophobic interactions, the hollow portion of the center cavity may hold a large number of hydrophobic guest molecules [[35]]. Antibiotics [[36]], antimicrobial peptides [[37]], photosensitizing agents [[38]], metallic nanoparticles [[39]], various autoinducers [[40]], essential oils [[41]], and other hydrophobic guests can all be included in cyclodextrins. While permeability and dissolution are both rate-limiting factors for BCS Class IV drugs, the dissolution phase is the rate-limiting element for the absorption of BCS Class II drugs [[42]]. Developing inclusion complexes with cyclodextrins can considerably enhance the water solubility of poorly soluble drugs [[43]]. In photodynamic treatment (PDT), a chromophore known as a photosensitizer (PS) is utilized to create highly reactive oxygen species (ROS), such as hydroxyl radicals (Type I photo-process) and cytotoxic singlet oxygen, by “sensitizing” the surrounding triplet oxygen upon light absorption (Type II photo-process) [44, 45, 46, 47].
The rise in antibiotic resistance has shifted the scientific community’s attention to using PDT to treat infections [[48]]. Due to excellent benefits such as the ease of application, the non-invasive nature of light, and, most importantly, the lack of potential ROS resistance in microbes, antimicrobial PDT (aPDT) has emerged as a major replacement for conventional antibiotic therapy [[49]]. Photodynamic therapy includes the systemic delivery of a photosensitizer, typically a porphyrin-based compound, which has been integrated into lipophilic drug carriers, such as liposomes, oil emulsions, or cyclodextrin inclusion complexes to reduce precipitation in the blood system or accumulation in a polar milieu and enhance PDT effectiveness [[50],[51]]. Targeting bacteria’s virulence factors, biofilm development, and protease and siderophore synthesis appear to be a more promising alternative strategy than focusing on them with bactericidal or bacteriostatic drugs. Such virulence factors as regulating virulence expression and bacterial adherence are necessary for infection [52, 53, 54]. Quorum sensing is a technique of cell-to-cell communication used by bacteria. Gram-negative bacteria often employ an acylated homoserine lactone (AHL) known as an autoinducer during quorum sensing. By trapping AHLs, cyclodextrin reduces quorum sensing [[55]]. Cyclodextrin nanocarrier also increases the QSI’s (Quorum sensing inhibitor) efficacy by improving the solubility and bioavailability [[56]]. In addition to enhancing medication permeability across the membrane barrier when employed as complexing agents, CDs may make antibiotics or other antibacterial agents more soluble. It increases the guest molecule’s bioavailability and alters the antibacterial activity and chemical stability [[57]]. For several bacterial strains, CD/antibiotic complexes displayed lower MIC values than antibiotics alone because CD or CD derivatives can increase the stability and permeability of antibiotics (Table 1) [[58]]. Frequent dosing of many antibiotics with short half-lives in conventional formulations is required to maintain antibacterial action. Otherwise, a concentration below the minimum inhibitory concentration (MIC) usually happens during anti-infective therapy, which results in antibiotic resistance.
By sustaining a consistent plasma drug concentration over MIC over a prolonged length of time, extended-release dosage forms improve the therapeutic effectiveness of antibiotics while decreasing antibiotic resistance. Improved patient compliance is an unquestionable benefit of extended-release formulations [[59]]. Due to its unique glucose ring topologies and many hydroxyl groups with a hydrophobic interior chamber and a hydrophilic exterior [[62]], cyclodextrins have been widely employed in creating medical devices to provide prolonged antibiotic-release properties [[60],[61]]. Antimicrobial medications can be captured by CD and released gradually at the necessary times [[63]]. We briefly address developments in the creation of cyclodextrin-based antibacterial agents in this review. The antibacterial activities of these supramolecular materials can be related to either the modified structure of macrocycles or the encapsulated antibiotics or other antibacterial agents in the cavities of these macrocycles, which further aid in activity enhancement for antimicrobials, decrease antibiotic resistance, disrupt biofilms, and minimize the chance of infection due to the host-guest complexation properties and easy and versatile functionality of cyclodextrin.
3.1 Cyclodextrin
By using bacterial digestion, Antoine Villiers of France extracted potato starch in 1891. He called this experimental substance dextrin “cellulose” [[131]]. Franz Schardinger found the crystalline molecules α-dextrin and β-dextrin in 1903, and Villiers’ “cellulose” were identified as β-dextrin. Today, these compounds are also known as α-cyclodextrin (α-CD) and β-cyclodextrin (β-CD) [[132],[133]]. Freudenberg et al. discovered a different substance in 1935 that they called γ-cyclodextrin (CD) [[134]]. CDs can create supramolecular structures called inclusion complexes (ICs) when combined with other molecules, primarily small molecules. CDs are truncated cone-shaped molecules with an external hydrophilic shell and a hydrophobic core (Fig. 3). The driving forces of IC (inclusion complex) formation is influenced by interactions, including hydrophobic, van der Waals, and hydrogen bonding, which can change guest molecules’ physical, chemical, and biological properties [[135],[136]]. CDs are regarded as effective drug-carrying and drug-delivering devices due to their capacity to create ICs. CDs are also effective solubilizers, physical stabilizers, and protectors of guest molecules against gastrointestinal tract degradation, increasing medication bioavailability [[137]]. Piroxicam/β-CD (Brexin® tablets) was the first pharmaceutical formulation to be released in Europe in 1977, and itraconazole/2-hydroxypropyl-βCD oral solution (Sporanox®) was the first to get US approval [[138]].
3.1.1 α-Cyclodextrin (α-CD)
CDs are primarily categorized according to how many glucose units are in their structure; for example, molecules with six glucose units are called α-CDs (Fig. 4) [[139]]. There is weak hydrogen bonding on the outer edge between the 2-OH and 3-OH groups; this interaction is weak in α-CD and highest in γ-CD. While 2-OH serves as the acceptor and 3-OH as the donor in α-CD, the bonding in β- and γ-CD alternates between 3-OH (acceptor) and 2-OH (donor). CDs are amphipathic structures with 6-OH groups on the narrower rim and 3-OH and 2-OH groups on the broader rim. The hydrophilic groups surround the molecular cavity of cyclodextrins in contrast with the hydrophobic interior coated by ether-like anomeric oxygen atoms. Due to its inadequate size, drugs cannot be contained in the cavity of α-CD [140, 141, 142].
3.1.2 β-Cyclodextrin (β-CD)
β-CD has frequently been used in the beginning phases of pharmaceutical applications because it is easily accessible and has a cavity size suitable for various pharmaceuticals [[142]]. Comparatively to other CDs, the cavity size of the β-CD is more suited to encapsulate a variety of compounds [[143]]. It is possible to employ β-CD to improve medication solubility, bioavailability, safety, and stability and as a carrier in drug formulation by forming inclusion complexes with drug molecules that are chemically bonded [[144]]. Hydrophilic, hydrophobic, and ionizable derivatives are the three classes into which β-CD derivatives are divided. Every group carries out a distinct task. Examples of hydrophilic CDs that can enhance the water solubility of weakly water-soluble molecules include 2,6-dimethyl-β-CD, 2,3,6, trimethyl-β-CD, 2-hydroxypropyl-β-CD (HP-β-CD), and maltosyl-β-CD. Similarly, CDs belonging to the hydrophobic group, such as 2,6-diethyl-β-CD, can slow down the dissolution rate of poorly water-soluble drugs [[145]]. Sulfobutyl-ether-β-cyclodextrin (SBECD), also known by the brand name Captisol®, carboxymethyl-β-cyclodextrin (CMCD), and hydroxypropyl-β-cyclodextrin (HPβCD) derivatives have all been used in drug formulation, especially for Class 2 and 4 drugs (low solubility and low permeability) [[52]], because they have a higher water solubility and better biocompatibility.
3.1.3 γ-Cyclodextrin (γ-CD)
Large internal cavity sizes and the ability to enclose larger molecules give the γ-CD a distinct advantage over the α- and β-CDs [[146]]. Compared to other natural cyclodextrins, γ-CD has the maximum water solubility due to its flexible and noncoplanar structure. This feature makes it a suitable host for the solubility improvement of less water-soluble drugs, further explaining its applicability in other industries [[147]]. Due to its difficulty passing across biological membranes, quick digestion in the gastrointestinal system, and unchanged excretion through the urine following parental delivery, γ-CD has a limited bioavailability [148, 149, 150, 151, 152]. The γ-CD is a superior choice to improve drug properties due to its high water solubility, greater cavity size, and most favorable toxicological profile [[146]].
Download the full article as PDF here Cyclodextrin: A prospective nanocarrier for the delivery of antibacterial agents against bacteria that are resistant to antibiotics
or read it here
Cyclodextrin: A prospective nanocarrier for the delivery of antibacterial agents against bacteria that are resistant to antibiotics, Pranoy Saha, Md Rajdoula Rafe, Open AccessPublished:August 23, DOI:https://doi.org/10.1016/j.heliyon.2023.e19287







