- Suppliers






ADM 
Antares 
Armor Pharma 
Asahi KASEI 
Ashland 
BASF 
Beneo 
Captisol 
Clariant 
Croda 
DFE Pharma 
Excipio Chemicals 
Fuji Chemical 
Gattefossé 
Gangwal 
IOI Oleo 
Ingredient Pharm 
JRS Pharma 
Kerry 
KLK Oleo 
Lipoid 
Lubrizol Life Science Health 
MAGNESIA 
MEGGLE 
Nagase Viita 
Nordic Bioproducts 
pharm-a-spheres 
PMC Isochem 
Seppic 
ShinEtsu 
Sigachi 
SPI Pharma 
Südzucker 
Vikram Thermo 
Zerion Pharma  All4Nutra
All4Nutra
- Suppliers
ADM Antares Armor Pharma Asahi KASEI Ashland BASF Beneo Captisol Clariant Croda DFE Pharma Excipio Chemicals Fuji Chemical Gattefossé Gangwal IOI Oleo Ingredient Pharm JRS Pharma Kerry KLK Oleo Lipoid Lubrizol Life Science Health MAGNESIA MEGGLE Nagase Viita Nordic Bioproducts pharm-a-spheres PMC Isochem Seppic ShinEtsu Sigachi SPI Pharma Südzucker Vikram Thermo Zerion Pharma - All4Nutra

A Mini-Review on Enhancing Solubility in Topical Hydrogel Formulations Using Solid Dispersion Technology for Poorly Water-Soluble Drugs

A Mini-Review on Enhancing Solubility in Topical Hydrogel Formulations Using Solid Dispersion Technology for Poorly Water-Soluble Drugs
Abstract
The solubility behavior of drugs is a critical factor in formulation development. Approximately 40–45% of new drugs face market entry challenges due to low water solubility. Enhancing drug bioavailability is thus essential in developing pharmaceutical dosage forms. Many biopharmaceutical class II and IV drugs are commonly prescribed to treat inflammations, infections, and pain from various pathologies. Their oral administration has several drawbacks, including significant first-pass liver effects, low bioavailability, and adverse gastrointestinal effects. Topical application has gained relevance due to its advantages in delivering drugs directly to the target site, avoiding gastrointestinal irritation, and increasing their effectiveness. However, topical hydrogel formulations with poorly water-soluble drugs face challenges related to the skin’s permeability. Therefore, preparing topical hydrogels using solid dispersions (SDs) is an effective strategy to enhance the dissolution rate of poorly soluble drugs, thereby improving their topical bioavailability. In this review, the concepts of SDs, topical delivery systems, and topical hydrogel formulations incorporating SDs, as well as their preparation methods, characterization, and applications, will be discussed.
1. Introduction
The solubility of a drug is a physicochemical property that influences its absorption and therapeutic efficacy. Drugs having poor aqueous solubility can lead to unsuccessful formulation development. Improving the solubility and dissolution rates of hydrophobic drugs remains a significant challenge in pharmaceutical development. Several strategies have been developed to enhance the solubility and bioavailability of poorly water-soluble drugs, including solid dispersions (SDs) [1,2,3], complexation [3], and micronization [4,5]. Lipid-based systems, such as self-emulsifying drug delivery systems (SEDDSs), which form fine emulsions in the gastrointestinal tract, have also shown promise in improving drugs’ solubility and absorption [6,7]. Other techniques like co-crystals [8,9] and nanonization [10,11] have been widely explored for their ability to enhance drug dissolution rates. Furthermore, nanostructured lipid carriers (NLCs) and solid lipid nanoparticles (SLNs) are emerging as effective carriers that offer improved drug stability, controlled release, and enhanced solubility [12]. Thiomers, a class of thiolated polymers, also contribute to improving bioavailability by prolonging the retention of drugs at the mucosal sites through bioadhesion mechanisms [13]. These diverse approaches are critical to overcoming the challenges associated with delivering poorly soluble drugs.
SDs have been shown to be an effective method for increasing the dissolution rate and bioavailability of a variety of poorly water-soluble active pharmaceutical ingredients (APIs). SDs refer to a class of solid products that have at least two separate components, in general a hydrophobic drug and a hydrophilic matrix. This matrix can either be amorphous or crystalline. There are several ways to prepare SDs, including spray-drying, gel entrapment, solvent evaporation, kneading, melting, a melting solvent method, co-precipitation, using modified solvents, co-precipitation with supercritical fluid and co-grinding, and evaporation [14].
The Biopharmaceutical Classification System (BCS) is a scientific framework that classifies drugs into four categories based on their aqueous solubility and intestinal permeability. This framework helps with predicting and deciding whether there is a need for in vivo bioequivalence studies. Drugs are divided into class I (high solubility, high permeability), class II (low solubility, high permeability), class III (high solubility, low permeability), and class IV (low solubility, low permeability). Both the World Health Organization (WHO) [15] and the U.S. Food and Drug Administration (FDA) [16] apply the BCS to support biowaivers, particularly for immediate-release oral dosage forms, streamlining the approval of generic medicines when certain criteria are met. Biopharmaceutical class II drugs, which have high permeability but low solubility, along with class IV drugs, which exhibit both poor solubility and permeability, are commonly prescribed to treat inflammations, infections, and pains caused by a variety of symptoms. However, their oral administration has many adverse effects, with the majority being related to the gastrointestinal tract [17], and since a large number of inflammatory infections occur locally and near the body’s surface [18], topical application has become more important, with the advantage of delivering drugs directly to the site of inflammation, avoiding gastrointestinal irritation and decreasing adverse systemic effects. Moreover, cutaneous inflammatory illnesses impact a high number of people worldwide. While these conditions generally have lower mortality rates compared to those for other diseases, they significantly affect patients’ quality of life. According to the Institute of Health Metrics and Dermatology Experts, dermatitis, cellulitis, psoriasis, acne vulgaris, seborrheic dermatitis, and pyoderma are among the most common skin diseases [19]. Hydrogels have emerged as versatile carriers for drug delivery due to their ability to retain large amounts of water while maintaining their structural integrity. Their biocompatibility and tunable properties make them particularly valuable for biomedical applications, including controlled drug release. Although traditionally used for hydrophilic drugs, recent advances have enabled the incorporation of hydrophobic drugs into hydrogel matrices, significantly enhancing their solubility, bioavailability, and controlled-release profiles. Several approaches have been developed to achieve this, including micelle formation, nanostructured lipid carriers, cyclodextrin inclusion complexes, and amphiphilic copolymers that create hydrophobic domains within the hydrogel network.
Thermosensitive hydrogels, such as those based on a poly(lactic-co-glycolic acid)-poly(ethylene glycol)-poly(lactic-co-glycolic acid) (PLGA-PEG-PLGA) triblock copolymer, are particularly promising, as they transition from a liquid into a gel at body temperature, enabling localized and sustained drug delivery. This feature is particularly relevant in cancer therapy, where hydrogels loaded with hydrophobic chemotherapeutic agents have shown improved therapeutic efficacy while minimizing systemic toxicity. Furthermore, pH-sensitive hydrogels have been designed to provide controlled drug release in response to environmental pH variations, making them suitable for targeted delivery to specific tissues or diseased areas. This ability to improve the solubility and controlled release of hydrophobic drugs is highly relevant in the context of topical drug delivery, where achieving adequate solubility and skin permeability remains a major challenge [20].
In this work, the combination of SDs with hydrogels for topical drug delivery, solid dispersion hydrogels (SDHs), has been reviewed. Several relevant studies on SDHs for topical applications were selected, spanning from 2014 to 2024. The selection of this limited number of studies was due to the specificity of the topical application domain. However, through a detailed exploration of this topic, the need to emphasize the advantages and potential of SDHs has been identified. Therefore, the focus of this mini-review is to provide a concise overview of SDs, topical delivery systems, and SDHs as effective approaches to enhancing the dissolution rate of poorly soluble drugs in topical preparations. This review includes an examination of the different types of SDs, their preparation methods, and topical delivery systems, before delving into hydrogels that incorporate SDs. It highlights the challenges associated with the low water solubility of APIs and their permeability through the skin. Additionally, the concepts of SDHs, their methods of preparation, their characterization, and their applications in boosting drugs’ topical bioavailability are discussed.
2. Solid Dispersions (SDs)
Gibbs free energy is a thermodynamic measure that indicates the maximum amount of useful work a system can perform at a constant temperature and pressure. It depends on enthalpy, which is related to the total heat content of the system, and entropy, which measures the disorder or randomness of the particles. A change in the Gibbs free energy determines whether a process will occur spontaneously. If this change is negative, the process is spontaneous; if it is positive, the process is non-spontaneous; and if it is zero, the system is in equilibrium. In the context of SD formulations, which are solid products containing a hydrophobic drug dispersed in hydrophilic carriers, this concept is particularly relevant. SD formulations increase a drug’s solubility, surface area, and dissolution rate by altering its crystalline structure, thus reducing its Gibbs free energy. This reduction in its Gibbs energy contributes to the enhancement of the drug’s solubility and dissolution rate, making the process more thermodynamically favorable [21].
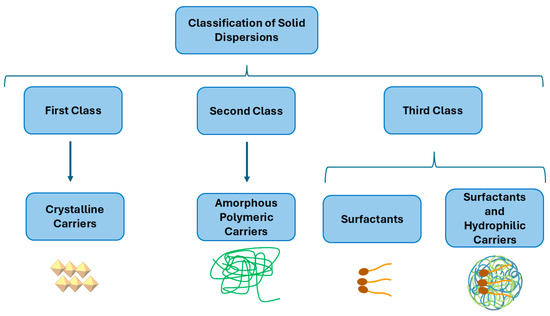
SDs are divided into three classes depending on the carriers that are used and on the fabrication method. The first class of SDs was created by Sekiguchi and Obi in 1961 [22]. In this class, an SD is produced utilizing crystalline carriers such as urea and sugars. However, these formulations were thermodynamically unstable, which resulted in slower drug release. Due to this thermodynamic instability of the first class of SDs [23], a second class of SDs was introduced that utilized amorphous polymeric carriers [24] instead of urea or sugars. These carriers are synthetic polymers such as polyethylene glycol (PEG) [25], polyvinylpyrrolidone (PVP) [26,27,28], polymethacrylates [29], natural polymers including ethyl cellulose [30], hydroxypropylmethylcellulose (HPMC) [31,32,33], and starch derivatives such as cyclodextrins (CDs) [34]. As a result, the drug particle size becomes smaller, improving its wettability and increasing its solubility.
Recently, a third class of SDs was created. A surfactant alone or in a combination with other hydrophilic carriers is used in the preparation of this class of SDs (Figure 1). This class provides a significant advantage in terms of its solubility, stability, and formulation flexibility, making these SDs a valuable tool in pharmaceutical development. Surfactants are commonly used to enhance the solubility of poorly water-soluble drugs and play a significant role in the pharmaceutical industry. Additionally, they serve as wetting agents, emulsifiers, detergents, dispersants, and foaming agents. Many surfactants are used to prepare SDs, such as inulin lauryl carbamate (Inutec®) [29], lauroyl polyoxyl-32 glycerides (Gelucire® 44/14 [35], glyceryl dibehenate (Compritol® 888 ATO) [36], a polyoxyethylene-polyoxypropylene block copolymer (Poloxamer 407) [37], and sodium dodecyl sulfate (SDS) [38].
Soluplus®-based solid dispersions have emerged as a promising strategy to enhance the solubility and bioavailability of poorly water-soluble drugs. Soluplus®, an amphiphilic polymer, facilitates the formation of stable amorphous solid dispersions, thereby improving drug dissolution rates. For instance, a recent study demonstrated that a Soluplus®-mediated amorphous solid dispersion of diosgenin (BCS class II) significantly increased its solubility and stability compared to these properties in the pure drug [39]. Additionally, third-generation solid dispersions combining Soluplus® and poloxamer 407 have been shown to enhance the oral bioavailability of resveratrol (BCS class II), increasing its solubility and reducing intestinal efflux and metabolism mechanisms [37]. These advantages underscore the potential of Soluplus®-based solid dispersions in improving the therapeutic performance of hydrophobic drugs.
SD formulations can be prepared using various methods, each with distinct advantages and limitations. These methods include solvent evaporation, kneading, melting, melting solvent methods, and ball milling (Figure 2).
The melting method, introduced by Sekiguchi and Obi in 1961 [40], involves heating a physical mixture of a drug and a hydrophilic carrier until both melt at a temperature slightly above their eutectic point. The resulting mixture is then rapidly cooled and solidified in an ice bath while being stirred. This approach is favored for its simplicity, cost-effectiveness, and ease of scalability. However, it poses a risk of thermal degradation for heat-sensitive drugs. Various fabrication techniques, such as hot melt extrusion and melt agglomeration, have been developed based on this principle, with continued use in modern research [41]. A notable variation of the melting method is the spray-congealing technique, which, although similar, introduces an additional atomization step. Instead of simply cooling the melted mixture, the spray-congealing method involves spraying it into a cold environment, causing the droplets to solidify into spherical particles. This modification allows for more controlled particle shapes and sizes, offering the potential to improve the solubility and bioavailability of poorly soluble drugs by enhancing their surface area. Both methods aim to enhance the solubility and bioavailability of poorly soluble drugs, but spray-congealing adds the step of atomization to create specific particle shapes and sizes [42].
The solvent evaporation method, which has been widely employed in the pharmaceutical industry since its introduction by Tachibana and Nakamura in 1965 [43], is especially useful for heat-sensitive compounds. In this process, both the drug and carrier are dissolved in a volatile solvent, eliminating the need for heat, which is a key advantage over the melting method. The solvent is then evaporated under constant stirring, leading to the formation of an SD. This technique is versatile and can be adapted through various fabrication processes, including spray-drying, electrospinning [44], lyophilization, supercritical fluid technology, and co-precipitation, which offer additional control over the final product’s properties [45].
The kneading method, studied by Dhandapani and El-Gied [29], involves dispersing the carrier in water to form a paste, into which the drug is incorporated and kneaded thoroughly. After kneading, the mixture is dried, and, if necessary, sieved. This method is advantageous because it is simple and scalable for industrial production and allows for the optimization of key process variables, such as the drug-to-polymer ratio and kneading time. Furthermore, it is compatible with a wide range of polymers, including surfactant-based ones like Poloxamer 188, which enhances the drug solubility and is generally safe for oral use [46]. However, it may not be as effective for drugs with extremely low solubility or for those that require fine-tuned particle size control.
The melting solvent method, as explored by Goldberg et al. [47], combines elements of both melting and solvent evaporation techniques. In this approach, the drug is dissolved in an appropriate solvent and then mixed with the melted carrier. The mixture is then evaporated to dryness, effectively creating an SD. This method is particularly beneficial for drugs with high melting points, although it requires careful control over the evaporation and mixing processes to avoid solvent residue or product inconsistency.
Lastly, the ball milling method involves grinding the drug and the carrier together in a ball mill to produce a uniform, fine mixture [48]. This technique is highly effective in enhancing the solubility and dissolution rate of poorly soluble drugs by increasing their surface area and reducing their particle size. While it offers clear benefits in terms of solubility enhancement, the ball milling method requires specialized equipment and may pose a risk of contamination from the milling media, which can impact the purity of the final product [49,50].
Each of these methods brings its own strengths and challenges, and the choice of method depends on the properties of the drug and the carrier and the desired characteristics of the final SD.
A recent study highlights the use of Fused Deposition Modeling (FDM) 3D printing technology as a novel approach to the fabrication of solid dispersions. This method offers precise control over the composition, structure, and release profile of drug delivery systems, making it a promising alternative to traditional fabrication techniques. FDM 3D printing enables the creation of solid dispersions with tailored physicochemical properties, such as a uniform drug distribution and enhanced dissolution rates. These properties are particularly beneficial for improving the solubility and bioavailability of poorly water-soluble drugs. This technology also provides advantages such as scalability, reproducibility, and the ability to design complex dosage forms, positioning it as an innovative and valuable tool for pharmaceutical development [51].
![Figure 2. Conventional manufacturing methods for SDs. This figure was created by the authors using BioRender.com (accessed on 5 March 2025) and Microsoft PowerPoint. The electrospinning illustration was adapted with modifications from Ref. [43], and the ball milling illustration was adapted with permission from Ref. [48]. Copyright 2021, Springer.](https://www.pharmaexcipients.com/wp-content/uploads/2025/03/figure.jpg)
While SDs offer numerous advantages for enhancing the bioavailability of drugs through an increase in their solubility, they also come with certain disadvantages, such as changes in crystallinity and a decline in the dissolution rate over time. Due to their thermodynamic instability, they are particularly sensitive to temperature and humidity during storage. These conditions can induce phase separation and crystallization by increasing the molecular mobility, lowering the glass transition temperature (Tg), or disrupting the interactions between the drug and its carrier, ultimately leading to the reduced solubility and dissolution rate of the drug [21,61]. To overcome this challenge, over the past few decades, various SD systems have been developed by incorporating poorly water-soluble drugs into different water-soluble carrier polymers. Numerous comprehensive reviews have summarized the methods for preparing, characterizing, and stabilizing these amorphous SDs [62,63]. To improve SDs and prevent the recrystallization of the API, carriers and surfactants can be optimized in several ways. Using polymers with high glass transition temperatures (Tg) helps maintain the API in an amorphous state, reducing the recrystallization risk. For example, PVP and HPMC are excellent carriers because they can form strong hydrogen bonds with the API, stabilizing the amorphous form. Eudragit® EPO, an amino methacrylate copolymer, is another effective carrier known for its ability to form stable hydrogen bonds with various drugs, further inhibiting recrystallization. Several factors, including reduced molecular mobility and drug–polymer interactions, have been found to prevent drugs’ crystallization from the amorphous state in polymer carriers. With the availability of a wide variety of polymers, a major challenge in SD design is the polymer selection.
Incorporating surfactants can enhance the miscibility of the drug and the carrier, stabilizing the amorphous form and increasing its solubility. Poloxamers (e.g., Poloxamer 188 and 407) are block copolymers that improve the solubility and dissolution rate of drugs by forming micelles and hydrogen bonds with the drug molecules. Gelucire® surfactants, such as Gelucire® 50/13 and 44/14, are lipid-based and help maintain the amorphous state of the drug by enhancing its miscibility with the carrier. SDS, an anionic surfactant, reduces surface tension and forms micelles, which can further stabilize the drug in its amorphous form. By carefully choosing and optimizing these carriers and surfactants, the stability and bioavailability of a drug can be significantly improved. The formation of hydrogen bonds between the drug and the carrier or surfactant is crucial, as these bonds reduce the mobility of the drug molecules, preventing them from rearranging into a crystalline structure and thus maintaining the drug in a more soluble and bioavailable amorphous state [21,64].
Download the full article as PDF here A Mini-Review on Enhancing Solubility in Topical Hydrogel Formulations Using Solid Dispersion Technology for Poorly Water-Soluble Drugs
or read it here
Materials
Table 1. Composition of SD formulations with drug-to-carrier ratios, their preparation methods, and solubility results.
| Preparation Methods | Drug: Carriers | Solubility (µg/mL or mg/mL) (Increase in Drug Solubility) |
Ref. | ||
|---|---|---|---|---|---|
| Melting | Indomethacin (IND):PEG 4000 IND:Gelucire® 50/13 |
Phosphate buffer, pH of 7.4 | El-Badry et al. [52] | ||
| 1:4 (4-fold) | |||||
| 1:4 (3.5-fold) | |||||
| Spray-drying | IND:PVP K25 | Phosphate buffer | Ji et al. [53] | ||
| pH of 4.98: 0.47 mg/mL | |||||
| pH of 6.05: 4.27 mg/mL | |||||
| pH of 7.25: 65.96 mg/mL | |||||
| Solvent evaporation | Flurbiprofen: Gelucire® 44/14 | Phosphate buffer solution, pH of 7.2, 0.618 ± 0.26 mg/mL (3.5-fold) |
Daravath et al. [54] | ||
| Solvent evaporation | IND:Eudragit® EPO (3:7) IND:HPMC (5:5 and 1:9) |
pH 2.2 Mcllvaine buffer > 80 µg/mL 36 µg/mL 35 µg/mL >80 µg/mL |
Xie et al. [55] | ||
| Spray congealing |
IND:Gelucire® 50/13: Gelucire® 48/16 (1:9:0) (1:4.5:4.5) (1:2.7:6.3) |
Phosphate buffer, pH of 5.8 0.194 ± 0.044 mg/mL (4-fold) 0.466 ± 0.045 mg/mL (19-fold) 0.775 ± 0.025 mg/mL (31-fold) |
Bertoni et al. [42] | ||
| Solvent evaporation | IND-Copovidone (IND-PVPVA) sodium IND–Copovidone (INDNa-PVPVA) |
Phosphate buffer, pH of 4.7–7.2 INDNa-PVPVA: 250 mg/mL |
Chiang et al. [56] | ||
| Lyophilization | IND:LHPC | Acetate buffer, pH of 5.8 656.09 ± 6.28 µg/mL (4.9-fold) |
Dahma et al. [38] | ||
| Lyophilization | Meloxicam:LHPC | Acetate buffer, pH of 5.8 709.17 ± 10.15 µg/mL (5.6-fold) |
Dahma et al. [57] | ||
| Lyophilization | Nystatin:Maltodextrin | Phosphate buffer, pH of 4.5 (1.3-fold) | Benavent et al. [58] | ||
| Ball milling | IND:Kaolin + 10% PVP+ 25% PVP+ 50% PVP+ 75% PVP |
Water 16.66 ± 0.1 µg/mL (1.8-fold) 29.05 ± 0.1 µg/mL (3.1-fold) 43.44 ± 0.1 µg/mL (4.5-fold) 44.44 ± 0.1 µg/mL (4.8-fold) |
Bejaoui et al. [49] | ||
| Ball milling | IND:lactose (1:1) | Water 39.18 ± 0.004 µg/mL (2.7-fold) |
Rojas-Oviedo et al. [50] | ||
| Solvent and melting | Kaempferol: Poloxamer 407 (1:5) |
Water 670.16 ± 90.53 µg/mL (4000 fold) |
Colombo et al. [59] | ||
Source: Dahma, Z.; Álvarez-Álvarez, C.; de la Torre-Iglesias, P.M. A Mini-Review on Enhancing Solubility in Topical Hydrogel Formulations Using Solid Dispersion Technology for Poorly Water-Soluble Drugs. Colloids Interfaces 2025, 9, 17. https://doi.org/10.3390/colloids9020017







